“ 空间转录组测序主要包括5个步骤,我们着重下游分析部分:空转数据分析和可视化。本篇主分享如何使用python和R读取空转数据,主要使用scanpy stlearn seurat包”
引言

在正式开始之前,我们先看看cellranger流程跑完之后,空间转录组结果的数据组成,主要是两部分:
-
1.表达矩阵文件夹
-
2.空转图片文件夹
-
1.表达矩阵文件夹

-
2.表达矩阵文件夹

-
另外:如果你对单细胞数据读取比较感兴趣,可以看我以前的贴子
-
1 Seurat读取不同数据格式以创建Seurat单细胞对象
-
2 GSE158055 covid19 肺组织60W单细胞细胞实战 wget -nd参数
代码实战
01
—
python中读取空间转录组的方式,scanpy较常见
-
1.1使用scanpy读取空转数据
在linux系统中的文件格式如下

import scanpy as sc#方法一ns7=sc.read_visium(path="./ns7/",count_file='./2.3.h5_files/filtered_feature_bc_matrix.h5',library_id="NS_7",load_images=True,source_image_path="./ns7/spatial/")#方法二adata=sc.read_visium(path="./ns56/",count_file='filtered_feature_bc_matrix.h5',library_id="NS_56",load_images=True,source_image_path="./ns56/spatial/")#读取之后,通常走一套组合拳,qc 可视化sc.pp.calculate_qc_metrics(adata, inplace=True)adata.var['mt'] = [gene.startswith('MT-') for gene in adata.var_names]adata.obs['mt_frac'] = adata[:, adata.var['mt']].X.sum(1).A.squeeze()/adata.obs['total_counts']fig, axs = plt.subplots(1,4, figsize=(15,4))fig.suptitle('Covariates for filtering')sb.distplot(adata.obs['total_counts'], kde=False, ax = axs[0])sb.distplot(adata.obs['total_counts'][adata.obs['total_counts']<10000], kde=False, bins=40, ax = axs[1])sb.distplot(adata.obs['n_genes_by_counts'], kde=False, bins=60, ax = axs[2])sb.distplot(adata.obs['n_genes_by_counts'][adata.obs['n_genes_by_counts']<4000], kde=False, bins=60, ax = axs[3])sc.pp.filter_cells(adata, min_counts = 5000)print(f'Number of cells after min count filter: {adata.n_obs}')sc.pp.filter_cells(adata, max_counts = 35000)print(f'Number of cells after max count filter: {adata.n_obs}')adata = adata[adata.obs['mt_frac'] < 0.2]print(f'Number of cells after MT filter: {adata.n_obs}')sc.pp.filter_cells(adata, min_genes = 3000)print(f'Number of cells after gene filter: {adata.n_obs}')sc.pp.filter_genes(adata, min_cells=10)print(f'Number of genes after cell filter: {adata.n_vars}')#这里最重要!!!!!!!!!sc.pp.normalize_total(adata, inplace = True)sc.pp.log1p(adata)sc.pp.highly_variable_genes(adata, flavor='seurat', n_top_genes=2000, inplace=True)sc.pp.pca(adata, n_comps=50, use_highly_variable=True, svd_solver='arpack')sc.pp.neighbors(adata)sc.tl.umap(adata)sc.tl.louvain(adata, key_added='clusters')#可视化sc.pl.umap(adata, color=['total_counts', 'n_genes_by_counts'])sc.pl.umap(adata, color='clusters', palette=sc.pl.palettes.default_20)sc.pl.spatial(adata, img_key = "hires",color=['total_counts', 'n_genes_by_counts'])sc.pl.spatial(adata, img_key = "hires", color="clusters", size=1.5)#详细内容可以参考https://scanpy-tutorials.readthedocs.io/en/multiomics/analysis-visualization-spatial.html



-
1.2 stlearn读取空转数据
https://stlearn.readthedocs.io/en/latest/tutorials/Read_any_data.html
需要三个文件:
-
count matrix
-
spatial location
-
image path (optional)
-
count

空间坐标信息

图片信息为可选
The image path is optional. If you don’t provide any image, the background will be 'black' or 'white'
import stlearn as stadata = st.create_stlearn(count=count_matrix,spatial=spatial,library_id="Sample_test", scale=1,background_color="white")
质控qc
st.pl.QC_plot(adata)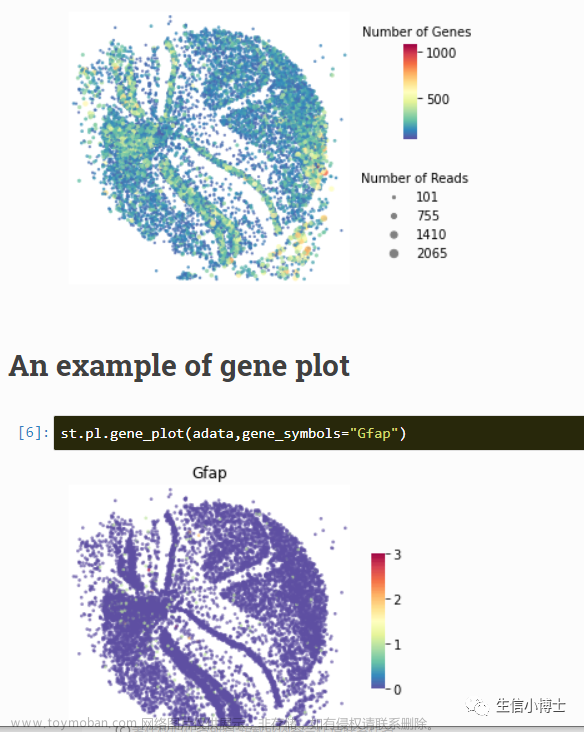
如果你还想添加metadata信息,可以添加seurat的聚类信息或者celltype
stLearn core object is using AnnData then you can transfer the clustering results to adata.obs. For example, you have clustering results from Seurat seurat_results (should be a numpy array), and then you can plot it with stlearn.pl.cluster_plot and use_label == "seurat"
adata.obs["seurat"] = pd.Categorical(seurat_results)02
—
R中读取空间转录组的方式,seurat最常见
-
2.1 如果你有h5文件+ spatial 文件夹
################################NS_7#首先读取表达矩阵数据#这里的blank是指使用空的spot,需要去掉。如果你没有空的spot,就不需要blankNS_7_exp <- Read10X_h5(filename = "/data/biomath/Spatial_Transcriptome/silicosis/Spatial_transcriptome/A72-NS-7d/2.3.h5_files/filtered_feature_bc_matrix.h5" ) #1638NS_7_blank <- as.matrix(read.csv("/data/biomath/Spatial_Transcriptome/silicosis/Spatial_transcriptome/A72-NS-7d/A72.csv",sep=",",header=T)) #283NS_7_valid=setdiff(colnames(NS_7_exp),NS_7_blank[,1])NS_7_exp_valid=NS_7_exp[,NS_7_valid] #1355NS_7=CreateSeuratObject(counts = NS_7_exp_valid, project = 'NS_7', assay = 'Spatial',min.cells=3) #17433*1355#读取image信息NS_7_img <- Read10X_Image(image.dir="/data/biomath/Spatial_Transcriptome/silicosis/Spatial_transcriptome/A72-NS-7d/spatial/")DefaultAssay(object = NS_7_img) <- 'Spatial'NS_7_img <- NS_7_img[colnames(NS_7)]NS_7[['image']] <- NS_7_imgNS_7$stim <- "NS_7"save(NS_7,file="NS_7.rds")#标准化 找高变基因 scale一条龙#通常空转数据使用sct效果比seurat标准流程更好NS_7_sct=SCTransform(NS_7, verbose = FALSE,,assay ="Spatial")save(NS_7_sct,file="NS_7_sct.rds")
-
2.2 如果你有raw/filtered_feature_bc_matrix 的三个文件 + spatial 文件夹
#首先读取表达矩阵数据NS2 = Read10X("./outs/filtered_feature_bc_matrix/")#读取image信息image2 <- Read10X_Image(image.dir = file.path("./outs/","spatial"), filter.matrix = TRUE)#这里如果你想要高清图片,可以指定的!image2 = Read10X_Image("./outs/spatial/",image.name = "tissue_hires_image.png")#创建seurat对象NS2 <- CreateSeuratObject(counts = NS2, assay = "Spatial")DefaultAssay(NS2 = image2) <- "Spatial"#标准化 找高变基因 scale一条龙#通常空转数据使用sct效果比seurat标准流程更好NS2_sct=SCTransform(NS2, verbose = FALSE,,assay ="Spatial")save(NS_7_sct,file="NS2_sct.rds")#如果多样本分析的时候记得改名字。image2 <- image2[Cells(x = NS2)]NS2[["slice1"]] <- image2#没有报错,无需转化.通常不会报错的for (i in colnames((NS2@images$slice1@coordinates))) {NS2@images$slice1@coordinates[[i]] <- as.integer(NS2@images$slice1@coordinates[[i]])}
-
2.3 一句话读取空转数据,这个通常是自己的空转数据可以这么读取。这是10x官网的数据
test_data = Load10X_Spatial(data.dir = "./input/",filename = "Visium_FFPE_Human_Normal_Prostate_filtered_feature_bc_matrix.h5",assay = "Spatial",slice = "test")
-
2.4 有的优秀作者会提供rds文件,这里主要注意版本问题,有时候用readRDS函数读取,有时候用load函数加载
pbmc=readRDS("./pbmc.rds")
load("./pbmc.rds")
-
2.5 非常规读取空转数据:有些奇葩作者不上传HE图片的,只给坐标信息
# 把空间数据当成单细胞数据读入#1 读取表达矩阵test_data2 = Read10X("./input/filtered_feature_bc_matrix/")test_data2 <- CreateSeuratObject(counts = test_data2,min.features = 0,project = "test")# 2读入单细胞的位置信息position = read.csv("./No_IHC/position_information.csv",header = T,row.names = 1)head(position)position = select(position,imagecol,imagerow)#将位置信息合并入单细胞seurat对象colnames(position) = paste0("Spatial_",1:ncol(position))test_data2[["spatial"]] <- CreateDimReducObject(embeddings = as.matrix(position),key = "Spatial",assay = "RNA")
-
2.6 非常规读取空转数据:有些奇葩作者不上传HE图片的,只给坐标信息,可采用Slide-seq的方法
test_data2 = Read10X("./input/filtered_feature_bc_matrix/")
test_data2 <- CreateSeuratObject(counts = test_data2,
min.features = 0,
project = "test",
assay="Spatial")
test_data2
# An object of class Seurat
# 17943 features across 2543 samples within 1 assay
# Active assay: RNA (17943 features, 0 variable features)
coord.df = read.csv("./position_information.csv",header = T,row.names = 1)
test_data2@images$image = new(
Class = 'SlideSeq',
assay = "Spatial",
key = "image_",
coordinates = coord.df
)后记
本文技术不难,更多的是一种思想方法,帮助大家去开拓自己的思路。只要你理解了空间数据的数据存储形式,便可一招鲜吃遍天~

生信小博士
【生物信息学】R语言,学习生信,seurat,单细胞测序,空间转录组。 Python,scanpy,cell2location,资料分享
公众号

 文章来源:https://www.toymoban.com/news/detail-830432.html
文章来源:https://www.toymoban.com/news/detail-830432.html
看完记得顺手点个“在看”哦!文章来源地址https://www.toymoban.com/news/detail-830432.html
到了这里,关于R、python读取空间转录组的8种方式的文章就介绍完了。如果您还想了解更多内容,请在右上角搜索TOY模板网以前的文章或继续浏览下面的相关文章,希望大家以后多多支持TOY模板网!